quarTeT: Telomere-to-telomere Toolkit
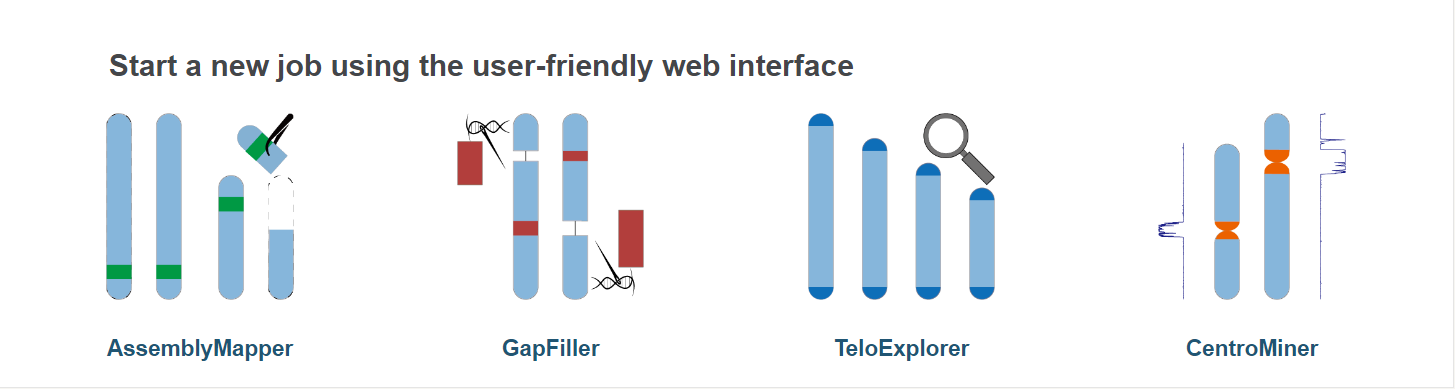
Task include:
- AssemblyMapper : reference-guided genome assembly
- GapFiller : long-reads based gap filling
- TeloExplorer : telomere identification
- CentroMiner : centromere candidate prediction
Getting Started
quarTeT can be easily accessed via our web server.
Go to home page, click corresponding module to start.
General
All modules have a similar structure.
Uploading data
If you access quarTeT from 443(https) port uploading may be slow.
Access quarTeT from 8080 port may accel uploading.
Setting algorithm parameters
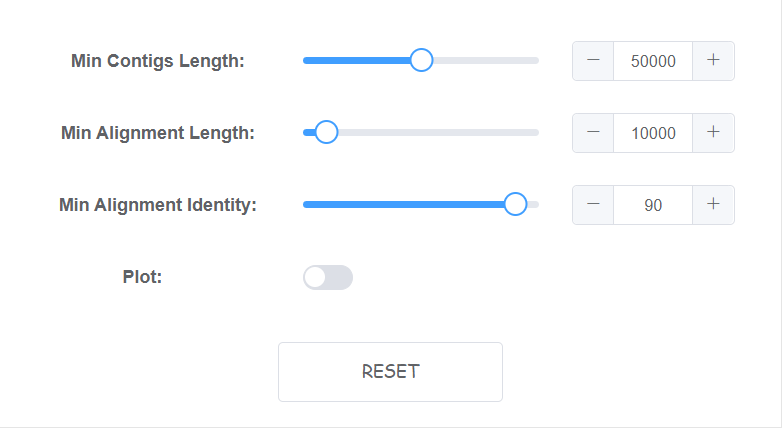
We have set default paramater which may solve most data.
If you are unsure of these parameter, just try default. You can click reset at the bottom right to get back default parameter.
Some paramater with bar can only switch in a window. However, you can directly input in box at the right. You will receive a warning if input is too large or small.
Submit task and access result
Click Run at the bottom right to submit task.
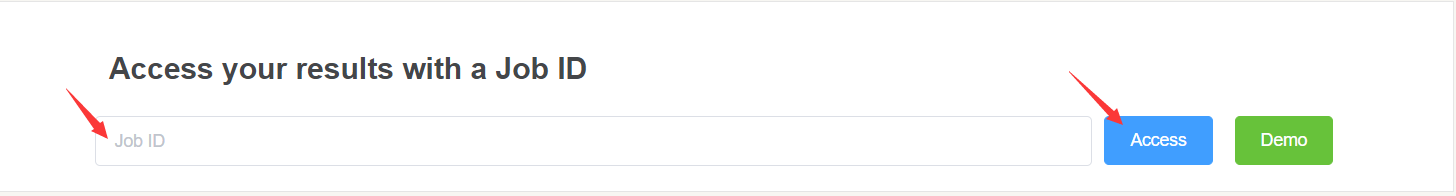
Once task is completed, we will email you the Job ID.
You can access your result from top of home page via Job ID.
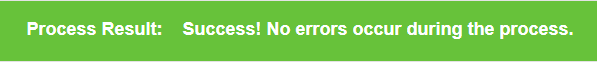
Some task may be processed with warning or error messages, you can check them at the top of result page.
Click download icon/buttom will open a new page. If download dialog doesn't pop up, right click the page and select Save as ... to download.
For plots, you can click it to enlarge.
For statistic tables, you can drag to resize tabs.
 AssemblyMapper
AssemblyMapper
AssemblyMapper is a reference-guided assemble tool.
Uploading sequence
You should upload 2 files in this section.
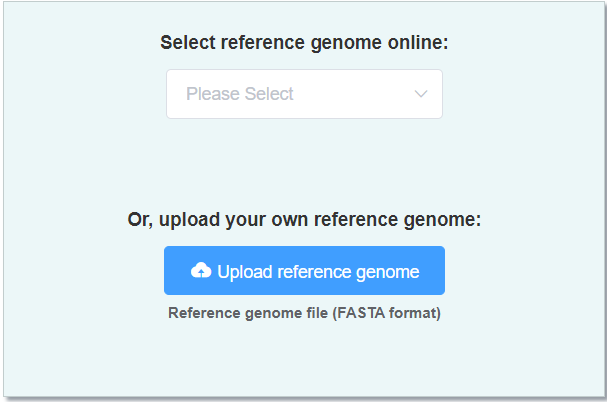
- Reference genome (FASTA format) a high-quality genome of close-related species. You can also select online reference genome here.
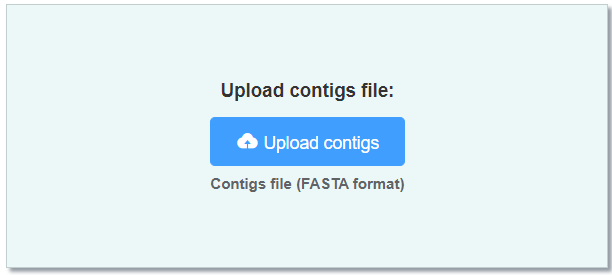
- Contigs (FASTA format) A phased contig-level assembly.
It's recommended to obtain such an assembly using hifiasm
you can convert {prefix}.bp.hap1.p_ctg.gfa and {prefix}.bp.hap2.p_ctg.gfa generated by hifiasm to FASTA format as input, separately.
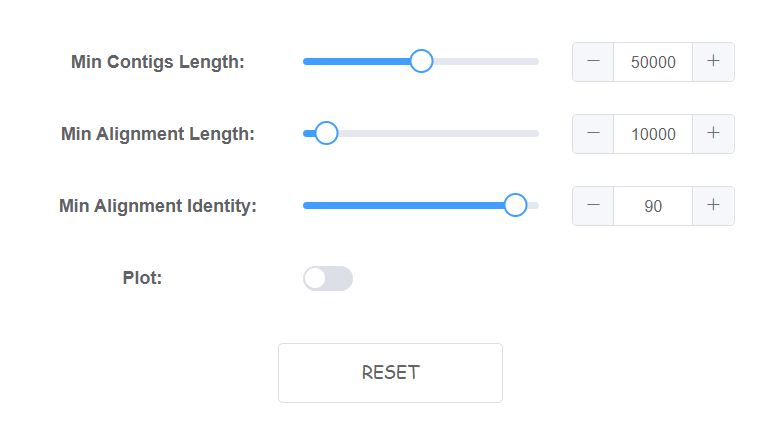
Setting algorithm parameters
4 parameters is availble on web for this module.
- Min Contigs Length Contigs shorter than INT (bp) will be removed, default: 50000.
- Min Alignment Length The min alignment length to be select (bp), default: 10000
- Min Alignment Identity The min alignment identity to be select (%), default: 90
- Plot Plot a colinearity graph for draft genome to reference alignments. (will cost more time)
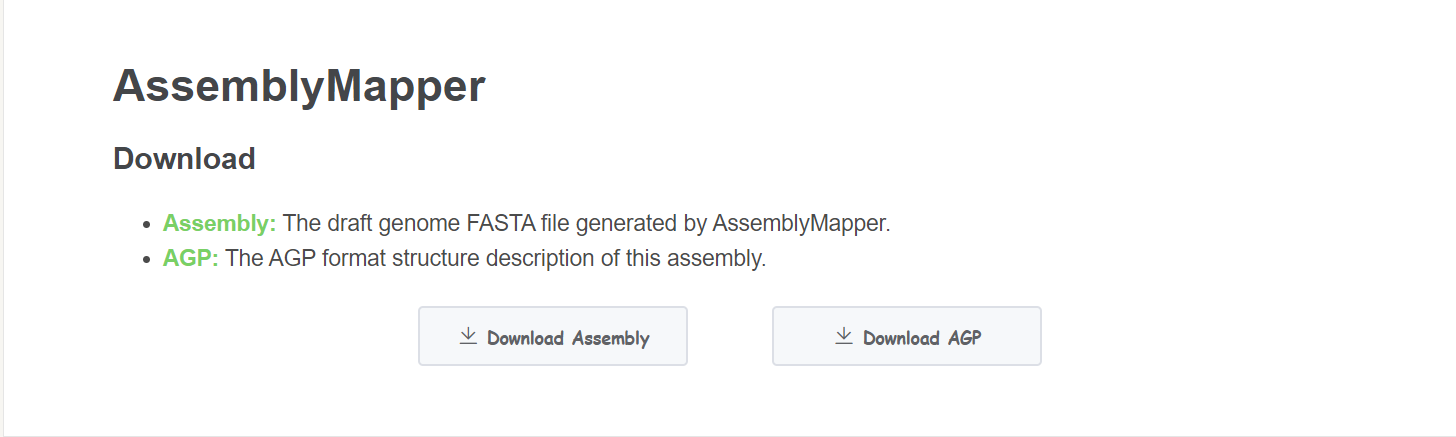
Result
- Download Click Download Assembly buttom to get the assembled genome. Click Download AGP to get the detailed assembly information.
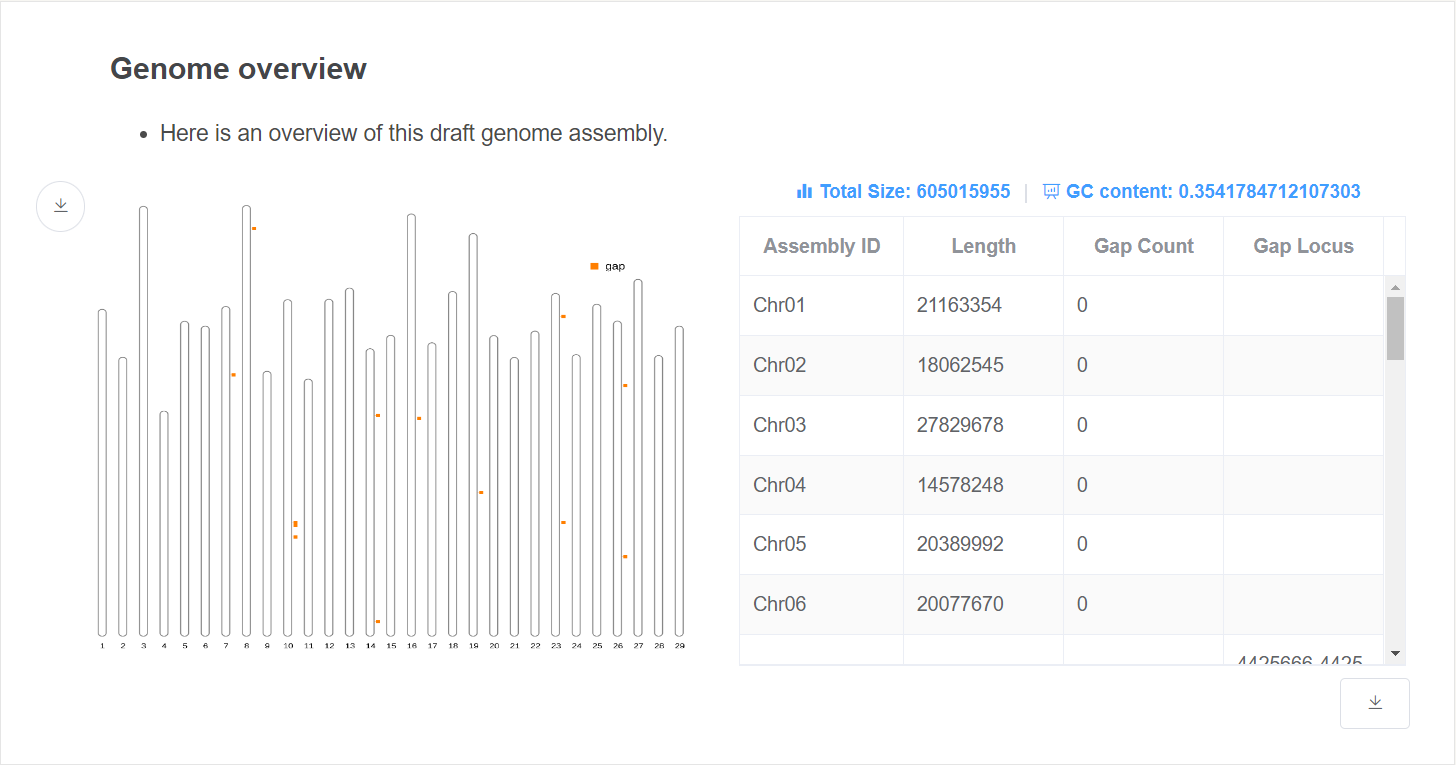
- Genome overview This plot gives an overview of the genome relative length and gap distribution.
- Genome statistic This table gives the length and gap location of each chromosome. Genome total size and GC content are given above the table.
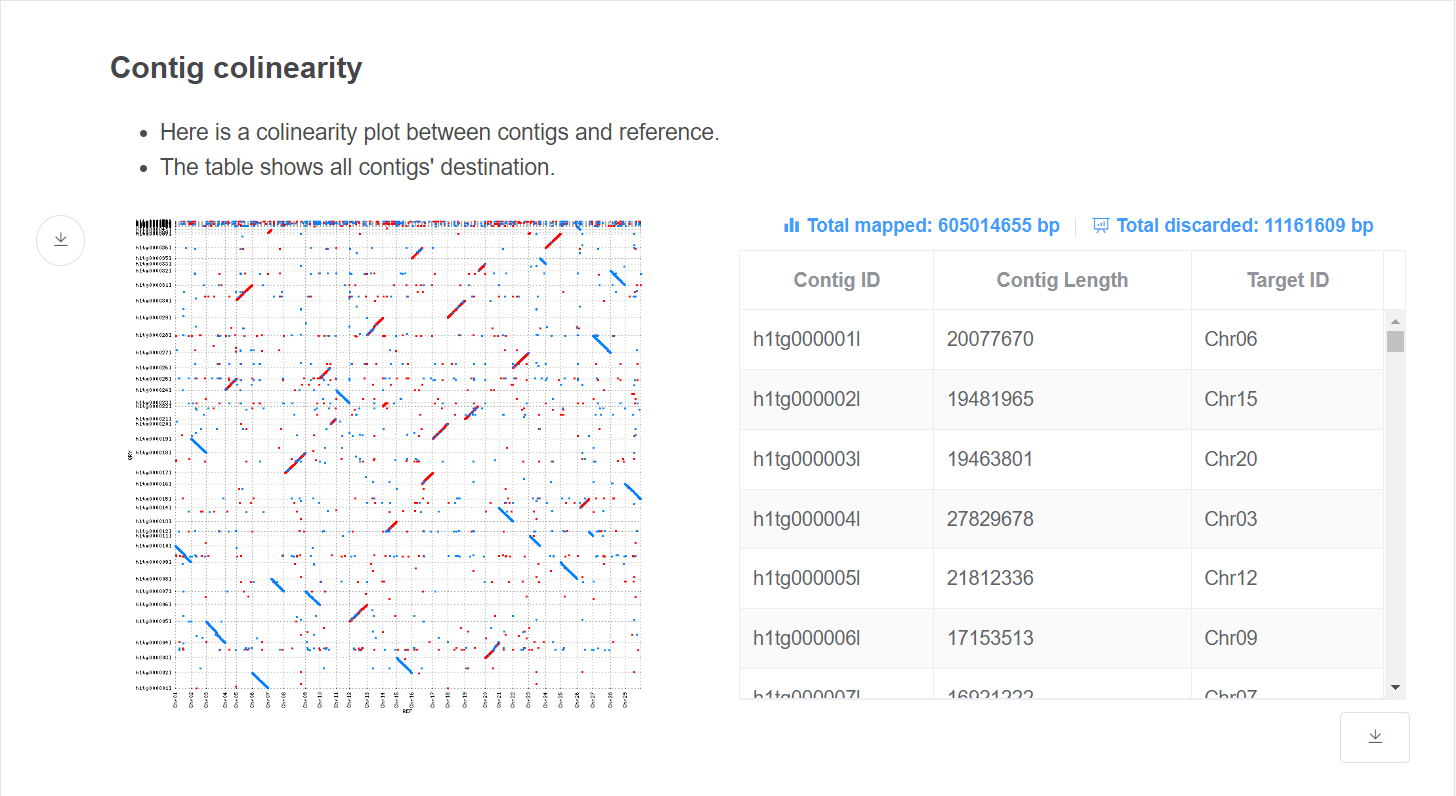
- Colinearity plot This plot gives colinearity information which decided contig placement.
- Alignment information This table gives the destination of each contig. Total discarded size are given above the table.
If Plot option is turned on, there will be another plot showing colinearity between assembled genome and reference.
 GapFiller
GapFiller
GapFiller is a long-reads based gap filling tool.
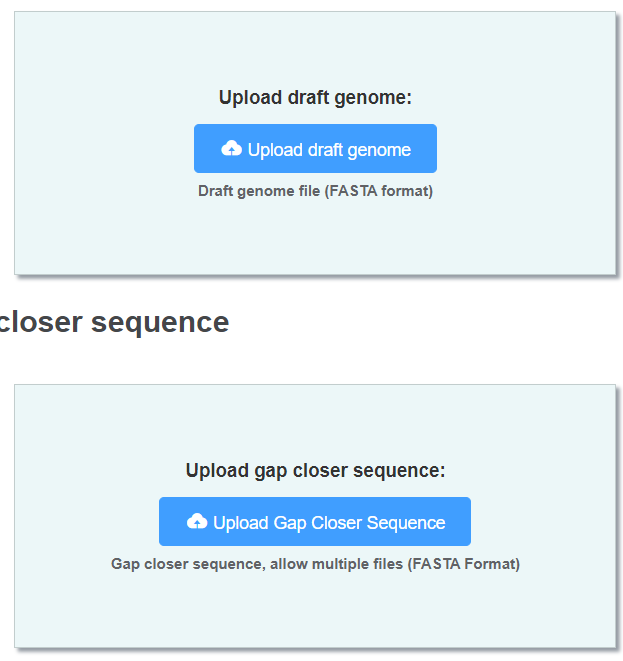
Uploading sequence
You should upload 2 files in this section.
- Genome (FASTA format) a gap-tied genome which you want to fill.
- Gap closer sequences (FASTA format, multiple files allowed) Sequence of the same genome generated by different platform or methods.
Long reads are acceptable, but contig-level assembly may achieve better result.
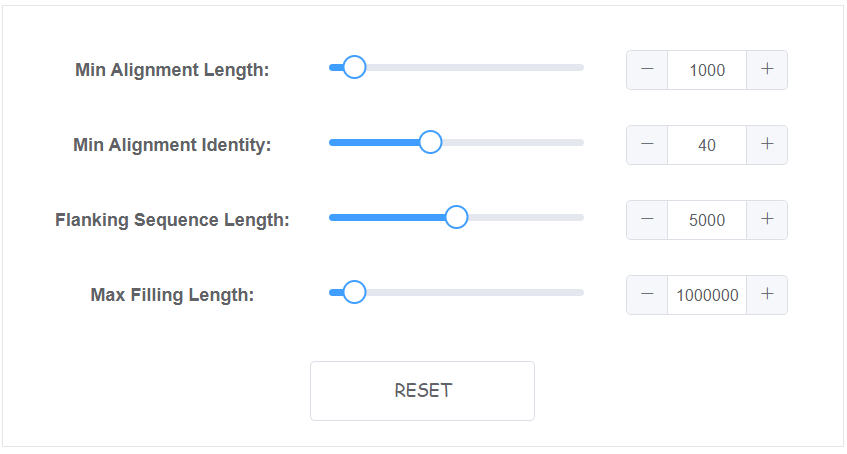
Setting algorithm parameters
4 parameters is availble on web for this module.
- Min Alignment Length The min alignment length to be select (bp), default: 1000
- Min Alignment Identity The min alignment identity to be select (%), default: 40
- Flanking Sequence Length The flanking sequence length besides gap which are used to anchor (bp), default: 5000
- Max Filling Length The max sequence length acceptable to fill any gaps, default: 1000000
Result
- Download Click Download Genome buttom to get the gap-filled genome.
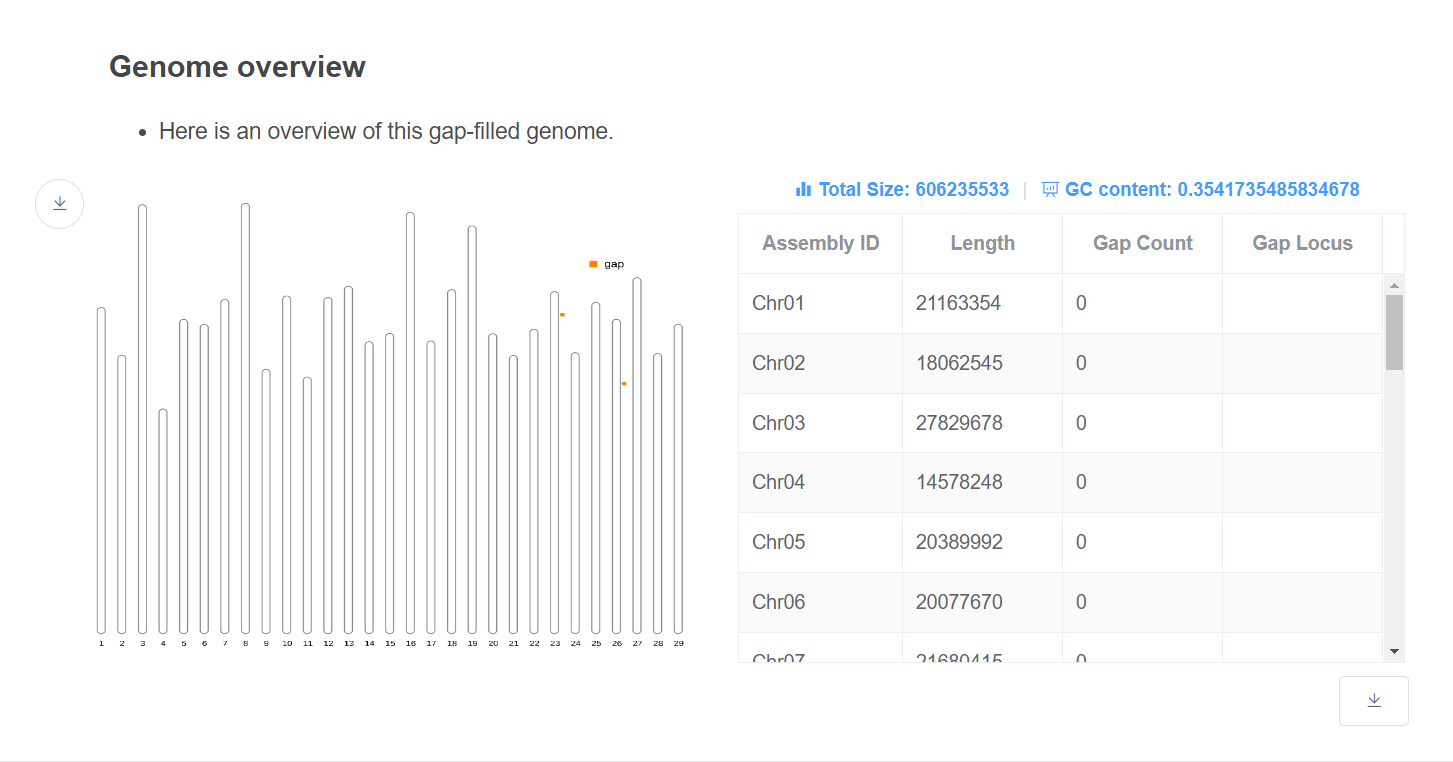
- Genome overview This plot gives an overview of the genome relative length and remaining gap distribution.
- Genome statistic This table gives the length and gap remaining location of each chromosome. Genome total size and GC content are given above the table.
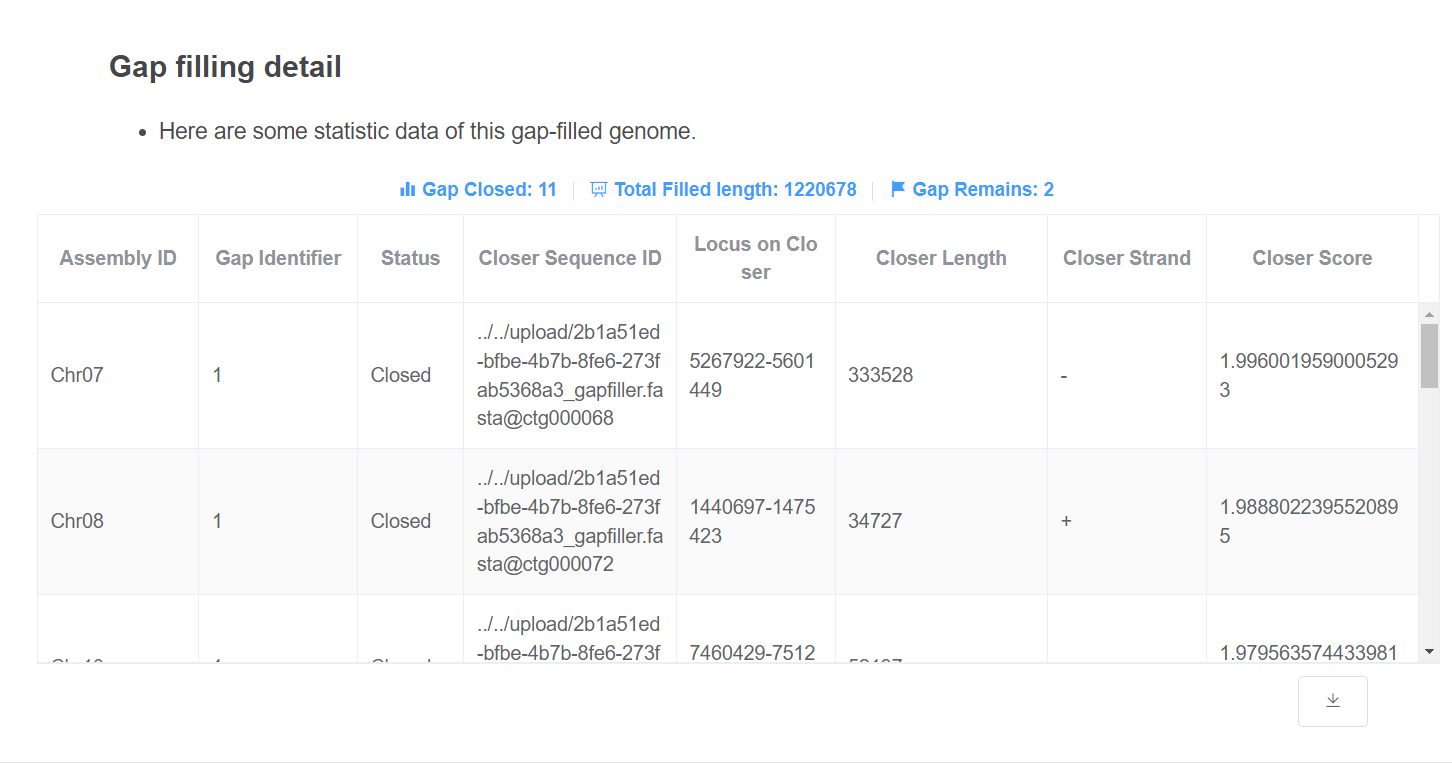
- Gap filling detail This table gives information about how the gaps are closed. Gap closed/remaining count and total filled length are given above the table.
 TeloExplorer
TeloExplorer
TeloExplorer is a telomere identification tool.
Uploading Genome
You should upload 1 file in this section.
- Genome (FASTA format) The genome you want to identify telomere.
Setting algorithm parameters
2 parameters is availble on web for this module.
- Min Repeat Times The minimal telomeric repeat times to be reported as telomere, default: 100
- Clade Specify clade of this genome. Plant will search TTTAGGG, animal will search TTAGGG, other will use tidk explore's suggestion, default: other
Result
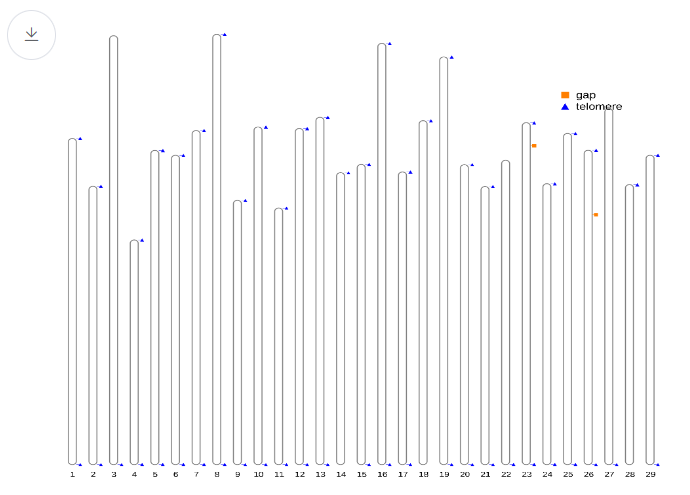
- Telomere overview This plot gives an overview of the genome relative length and telomere location alongside gap distribution.
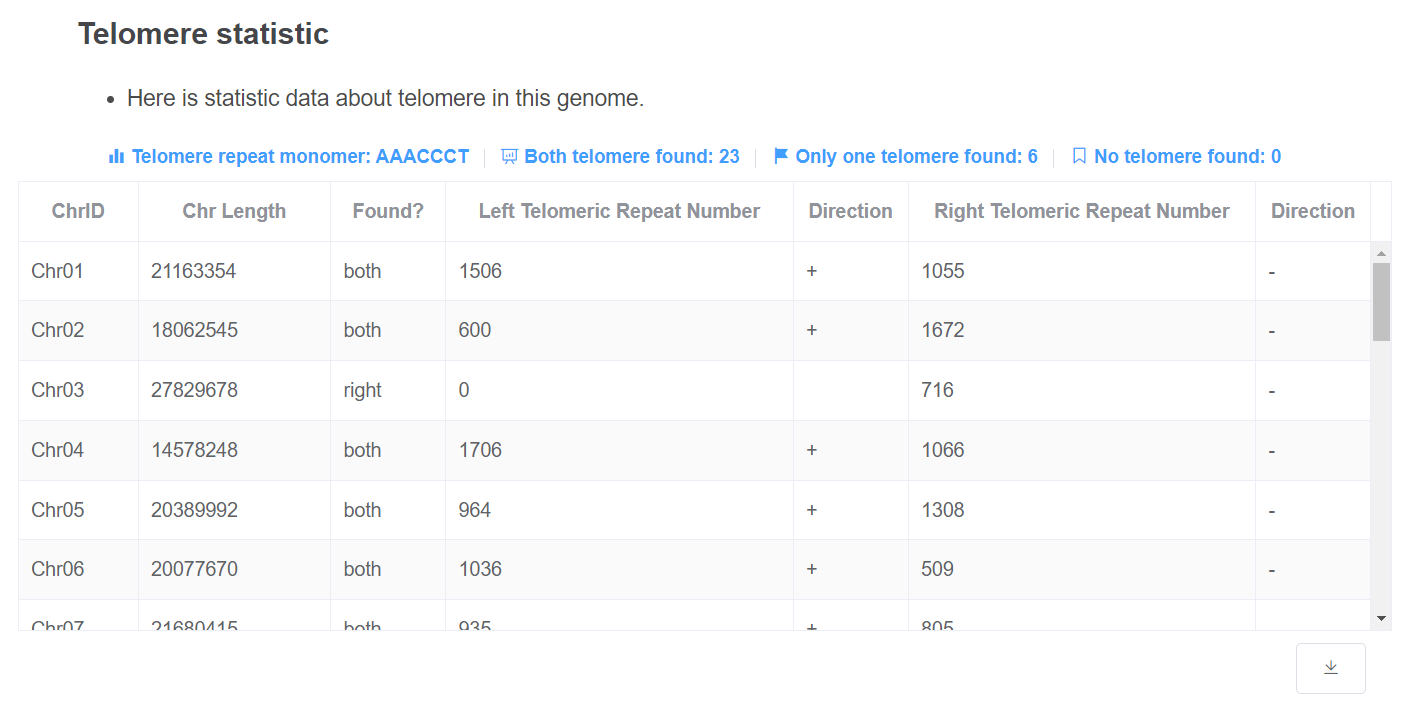
- Telomere statistic This table gives the telomere repeat times of each chromosome at each end. Telomere repeat monomer and total telomere found are given above the table.
 CentroMiner
CentroMiner
CentroMiner is a centromere prediction tool.
Uploading files
You should upload at least 1 file in this section.
- Genome (FASTA format) The genome you want to predict centromere.
- TE annotation (optional, GFF3 format) TE annotation, or just LTR annotation of this genome.
This file is optional. CentroMiner can run without it, but may improve performance with it.
It's recommended to obtain TE annotation using EDTA
{genome file}.mod.EDTA.TEanno.gff3 generated by EDTA can directly feed CentroMiner, unless you have sequence ID longer than 15 characters.
Note that the sequence ID in first column should be consistent with in genome. Some tools may change sequence ID if ID is too long.
The sequence ontology in the third column should include "LTR" to be recognized.
Setting algorithm parameters
3 parameters is availble on web for this module.
- Period Period (monomer length) range to be consider as centromere repeat monomer. Default: 100~200
- Max Gap Max allowed gap size between two tandem repeats to be considered as in one tandem repeat region. Default: 50000
- Min Length Min size of tandem repeat region to be selected as candidate. Default: 100000

Result
- Download Click Download candidates to get an archive of all centromere candidates information for each chromosome. This is the same as the table in Show More. Click Download TR annotation to get an archive of all tandem repeat annotation for each chromosome. Click Download TR sequence to get an archive of all tandem repeat monomer sequence.
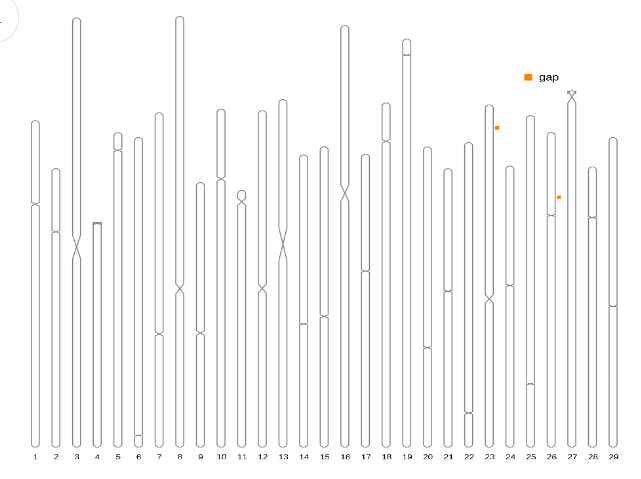
- Centromere overview This plot gives an overview of the genome relative length and predicted best centromere location alongside gap distribution.
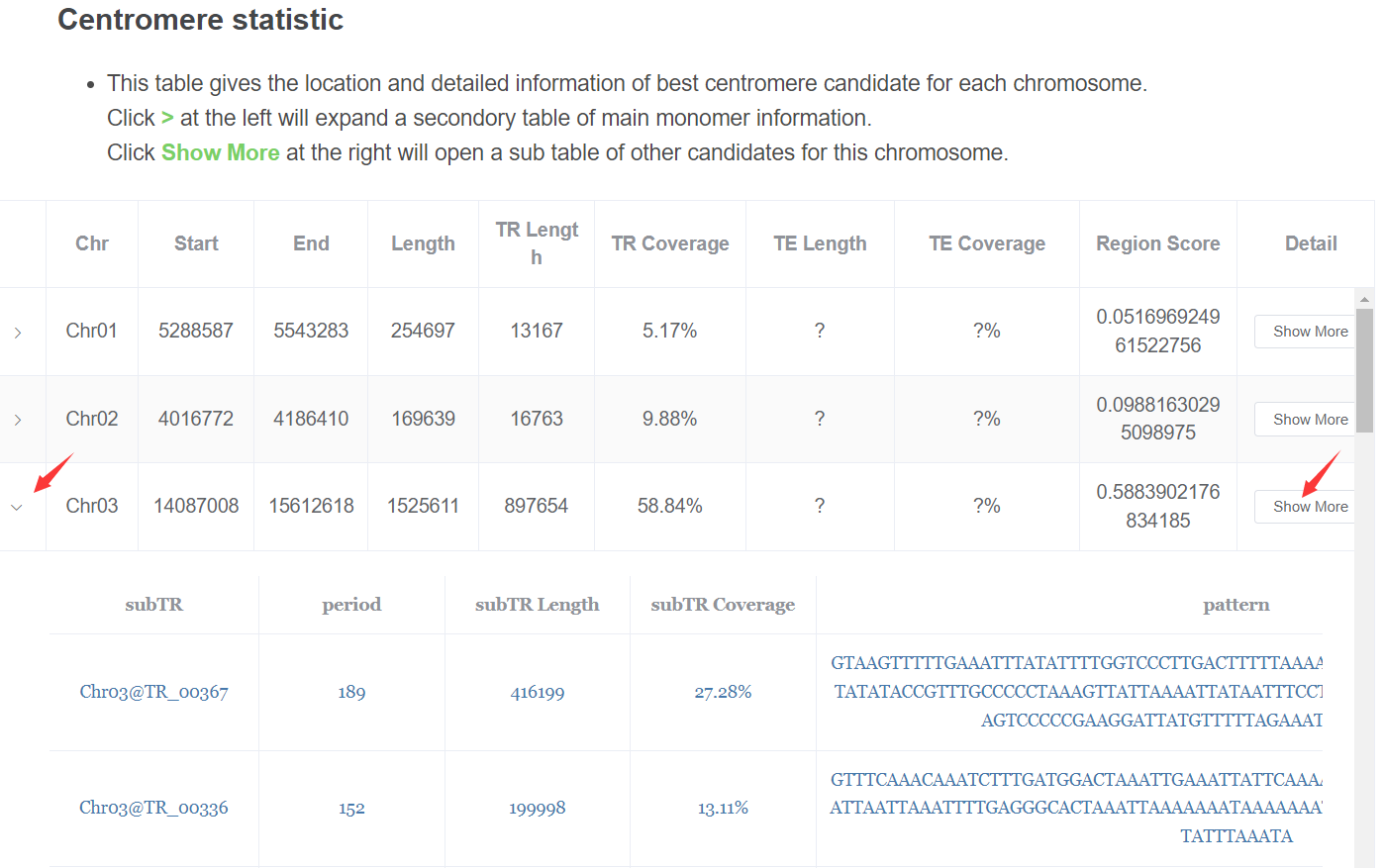
- Centromere candidates statistic This table gives the location and detailed information of best centromere candidate for each chromosome.
Click > at the left will expand a secondory table of main monomer information.
Click Show Moreat the right will open a sub table of other candidates for this chromosome.